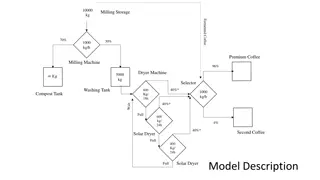
Genome.re-seq Data Processing Workflow

Create directories for error and output files, map read group information to a GATK approved genome, convert to sorted BAM, mark duplicates, and realign and base recalibrate using GATK programs. Includes instructions for script creation with SBATCH settings, file paths, tool usage, and memory limits.

Download Presentation

Please find below an Image/Link to download the presentation.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author.If you encounter any issues during the download, it is possible that the publisher has removed the file from their server.
You are allowed to download the files provided on this website for personal or commercial use, subject to the condition that they are used lawfully. All files are the property of their respective owners.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author.
E N D
Presentation Transcript
Make a new directory for error and out files Type: mkdir p ~/GATK/e_and_o/
The path Map with read group information to a GATK approved genome Convert to sorted bam sort by coordinates adjust qual scores re-align identify regions new bam files identify regions new bam files mark duplicates
Create a mapping script (GATK requires read groups) SBATCH settings #SBATCH --nodes=1 #SBATCH --ntasks=4# #SBATCH --time=00:10:00 # Time limit hrs:min:sec #SBATCH -p compute #SBATCH --mem=5gb # Memory limit We will use hisat2 and samtools, make sure they are in your path -x should be /scratch/Workshop/SR2019/8_GATK/ hg38_GATK/HISAT/genome The fastq files are in /scratch/Workshop/SR2019/8_GATK/reseq/ They are paired end! Convert to sorted bam (can you remember how?)
Create a script to mark duplicates and sort by coorinates SBATCH settings #SBATCH --nodes=1 #SBATCH --ntasks=4# #SBATCH --time=00:10:00 # Time limit hrs:min:sec #SBATCH -p compute #SBATCH --mem=5gb # Memory limit We will samtools, make sure it is in your path mkdir /scratch/Users/<your username>/tmp/ before you start! This uses the picard program SortSam and MarkDups bam files are input and output java -Xmx5G -Djava.io.tmpdir=/scratch/Users/<your username>/tmp/${SLURM_JOBID} -XX:ParallelGCThreads=4 -jar /opt/picard/2.6.0/picard-2.6.0.jar SortSam INPUT=<INFILE> OUTPUT=<OUTFILE> SORT_ORDER=coordinate java -Xmx5G -Djava.io.tmpdir=/scratch/Users/ <your username>/tmp/${SLURM_JOBID} -XX:ParallelGCThreads=4 -jar /opt/picard/2.6.0/picard-2.6.0.jar MarkDuplicates INPUT=<INFILE> OUTPUT=<OUTFILE> M=<MarkdupOUTFILE> After you mark duplicates you need to index the bam file you made as output Use samtools to index the bam files
Create a script to realign and base recalibrate SBATCH settings #SBATCH --nodes=1 #SBATCH --ntasks=4# #SBATCH --time=02:00:00 # Time limit hrs:min:sec #SBATCH -p compute #SBATCH --mem=5gb # Memory limit This uses the GATK programs RealignerTargetCreator (hint: needs all 4 bams in one command) IndelRealigner (on each bam) BaseRecalibrator (on each bam) PrintReads(on each bam)