Rejection of Tissue Transplants and Immune Responses

The immune mechanisms behind tissue rejection in transplants, focusing on allografts and effector mechanisms leading to graft failure. Exploring hyperacute rejection and immune recognition of allografts.

Uploaded on Mar 03, 2025 | 0 Views
Download Presentation

Please find below an Image/Link to download the presentation.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author.If you encounter any issues during the download, it is possible that the publisher has removed the file from their server.
You are allowed to download the files provided on this website for personal or commercial use, subject to the condition that they are used lawfully. All files are the property of their respective owners.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author.
E N D
Presentation Transcript
IMMUNOPATHOLOGY DR.MAYSEM LEC.4
Rejection of Tissue Rejection of Tissue Transplants: Transplants:
-Allografts transplantation of organs from one individual toanother of the same species. Rejection is a complex phenomenon involving both cell- and antibody-mediated reactions that destroy the graft.
Immune Recognition of Allografts Rejection of allografts is a response mainly to MHC molecules. There are two main mechanisms by which the host immune system recognizes and responds to the MHC molecules on the graft:
1.Direct pathway: Host T cells recognize donor HLA on antigen presenting cell s (APC) derived from the donor. 2.Indirect pathway: Host T cells recognize donor HLA after processing and presention on host APC (analogous to any other exogenous processed antigen).
Effector Mechanisms of Graft Rejection Direct cytotoxic T cell (CTL-) mediated parenchymal and endothelial cytolysis Macrophage-mediated damage Cytokine-mediated vascular and parenchymal dysfunction Microvascular injury also causes downstream tissue ischemia Antibody-mediated responses can also be important; these tend to induce injury to endothelial cells rather than parenchymal cells
Hyperacute rejection: Pre-formed antidonor antibodies bind to graft endothelium immediately after transplantation, leading to thrombosis, ischemic damage, and rapid graft failure
Hyperacute rejection occurs when the recipient has been previously sensitized to graft antigens (e.g., by blood transfusion or pregnancy). Preformed, circulating antibody binds to graft endothelial HLA with an immediate (minutes to days) complement- and antibody -mediated injury.
Acute Rejection Acute rejection typically occurs within days or months of transplantation or after cessation of immunosuppressive therapy. Both cellular and humoral mechanisms can contribute.
Acute cellular rejection T cells destroy graft parenchyma (and vessels) by cytotoxicity and inflammatory reactions. Acute humoral rejection (rejection vasculitis) is mediated by newly synthesized (not preformed) anti-donor antibodies thatcause a necrotizing vasculitis with consequent thrombosis.
Chronic Rejection Chronic rejection occurs over months to years and is characterized by progressive organ dysfunction. Dominated by arteriosclerosis, this type is probably caused by T cell reaction and secretion of cytokines that induce proliferation of vascular smooth muscle cells, associated with parenchymal fibrosis
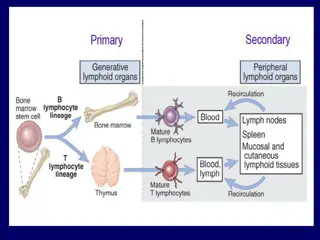
Transplantation of Hematopoietic Stem Cells(HSC) Hematopoietic stem cell (HSC) transplantation is used astherapy for: 1. hematopoietic malignancies.e.g. leukemia and some nonhematopoietic malignancies 2.aplastic anemias, 3. certain inherited disorders e.g. immune deficiency states and severe forms of thalassemia.
Sources of Hematopoietic Stem Cells for Transplantation: 1.from donor bone marrow, 2. from the peripheral blood. 3. from the umbilical cord blood of newborns, a readily available rich source of HSCs
The recipient receives chemotherapy and/or irradiation to destroy malignant cells (e.g., in leukemia) and to create a graft bed; then, HSCs are infused into the peripheral blood, from which they home to bone marrow .
Rejection of allogeneic HSC transplants seems to be mediated by some combination of host T cells and NK cells that are resistant to radiation therapy and chemotherapy.
Major problems complicate this form of transplantation: 1.graft-versus-host disease(GVHD) : This occurs when immunologically competent T cells (or their precursors) are transplanted into recipients who are immunologically compromised.
-Acute GVHD (occurring days to weeks after transplantation)causes epithelial cell necrosis in three principal target organs: liver, skin, and gut. -Chronic GVHD may follow the acute syndrome or mayoccur insidiously. The patients develop skin lesions resembling those of systemic seclerosis.
2. Immune Deficiencies. These are often of prolonged durationin recipients of HSC transplants. Recipients are susceptible to a variety of infections, mostly viral, such as cytomegalovirus (CMV) and EBV infections.
Amyloidosis Amyloidosis is a disorder characterized by the extracellulardeposits of misfolded proteins that aggregate to form insoluble fibrils. .
Pathogenesis of Amyloidosis : Normally, misfolded proteins are degraded intracellularly in proteasomes, or extracellularly by macrophages. It appears that in amyloidosis, these quality control mechanisms fail,
allowing the misfolded protein to accumulate outside cells. Misfolded proteins often are unstable leading to the formation of oligomers and fibrils that are deposited in tissues
Proteins that form amyloid are either: Normal proteins that have a propensity to fold improperly andassociate to form fibrils; over-production (or defective catabolism) thus leads to deposition. Mutant proteins that are prone to misfolding and aggregation; even normal levels of synthesis can cause deposition
Of the many biochemically distinct forms of amyloid proteins that have been identified, three are most common:
1 The AL (amyloid light chain) protein is produced by plasma cells and is made up of complete immunoglobulin light chainsor the amino-terminal fragments of light chains, or both. The deposition of amyloid fibril protein of the AL type is associated with some form of monoclonal B cell proliferation
2.The AA (amyloid-associated) fibril derived from a larger serum precursor called SAA (serum amyloid-associated) protein that is synthesized in the liver under the influence of cytokines that are produced during inflammation; thus, long-standing inflammation leads to elevated SAA levels, and ultimately the AA form of amyloid deposits
3.-Amyloid (A): A peptide that forms the core of cerebral plaques and deposits within cerebral vessel walls in Alzheimer disease; it derives from a transmembrane amyloid precursor protein
Other less-common forms of amyloid: Transthyretin (TTR): A normal serum protein that binds andtransports thyroxine and retinol. Excess amounts of normal TTR can deposit in hearts in senile systemic amyloidosis.
while mutant forms of the protein are deposited in a group ofhereditary diseases called familial amyloid polyneuropathy
2-microglobulin: component of class I HLA molecules and a normal serum protein; it is deposited in a form of amyloidosis that complicates long-term hemodialysis.
Classification of Amyloidosis Amyloid may be: 1.systemic (generalized), involving several organ systems, or 2.localized, when deposits are limited to a single organ, such as the heart.
On clinical grounds, the systemic, or generalized, pattern is subclassified into primary amyloidosis and secondary amyloidosis . Also there is Hereditary or familial amyloidosis
Primary amyloidosis: Immunocyte dyscrasias with amyloidosis is due to AL-type amyloid; it occurs in 5% to 15% of patients with multiple myeloma
Reactive secondary amyloidosis This is due to AA-type amyloid. Secondary amyloidosis is associated with chronic inflammatory states (e.g., rheumatoid arthritis, scleroderma, bronchiectasis, chronic osteomyelitis), and non-immunocyte tumors (e.g., Hodgkin lymphoma and renal cell carcinoma).
Hemodialysis-associated amyloidosis: In patients on chronic hemodialysis. -due to deposition (in joints, synovium, and tendon sheaths) of 2-microglobulin not filtered by normal dialysis membranes.
-Familial (Hereditary) Amyloidosis A variety of Rare familial forms of amyloidosis have been Described. The best-characterized is an autosomal recessive condition called familial Mediterranean fever
characterized by attacks of fever accompanied by inflammation of serosal surfaces, including peritoneum, pleura, and synovial membrane. This disorder is encountered largely in persons of Armenian, Sephardic Jewish, and Arabic origins .
- Localized Amyloidosis Sometimes deposition of amyloid is limited to a single organ or tissue without involvement of any other site in the body. .
The deposits may produce grossly detectable nodular masses or be evident only on microscopic examination. Nodular (tumor-forming) deposits of amyloid are most often encountered in the lung, larynx, skin, urinary bladder, tongue, and the region about the eye
Endocrine amyloid -occurs in tumors associated with hormone synthesis; for example, thyroid medullary carcinoma making procalcitonin that deposit as amyloid fibrils
Amyloid of aging occurs typically in the eighth and ninth decadesand is most commonly due to deposition of non- mutant transthyretin. Although amyloid distribution is systemic, the dominant involvement is of the heart.
Amyloid deposits cause tissue injury and impair normal function by causing pressure on cells and tissues. They donot evoke an inflammatory response.
The diagnosis of amyloidosis 1 -Biopsy and subsequent Congo red staining is the most important tool in the diagnosis of amyloidosis. In general, biopsy is taken from the organ suspected to be involvedRectal and gingival biopsy specimens contain amyloid in as many as 75% of cases with generalized amyloidosis
Haematoxylin and eosin-stained tissue : amorphous, acellular, hyaline, eosinophilic extracellular material -Congo red staining: amyloid is salmon-pink, which produces green birefringence under cross-polarized light and is the diagnostic gold standard.
2. Electrone microscope 3.In suspected cases of AL amyloidosis and immunoelectrophoresis should be performed. Bonemarrow examination in such cases usually shows plasmacytosis, 4.Proteomic analysis of affected tissue is now being widely used for detection of small amounts of amyloid (from fat aspirates) and for definitive identification of the type of amyloid.





























")
")
:")








 Amyloidosis")