Understanding Multiple Sequence Alignment with Hidden Markov Models

Multiple Sequence Alignment (MSA) is essential for various biological analyses like phylogeny estimation and selection quantification. Profile Hidden Markov Models (HMMs) play a crucial role in achieving accurate alignments. This process involves aligning unaligned sequences to create alignments with gaps if needed, ensuring accuracy by measuring parameters like Sum-of-pairs false negatives and false positives. Various methods such as MAFFT, MUSCLE, and newer approaches like SATe and PASTA are used for MSA, addressing challenges like sequence length heterogeneity. The use of profile HMMs allows for precise alignment through algorithms like Viterbi and Forward.

Download Presentation

Please find below an Image/Link to download the presentation.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author. Download presentation by click this link. If you encounter any issues during the download, it is possible that the publisher has removed the file from their server.
E N D
Presentation Transcript
Making A Multiple Sequence Alignment with Hidden Markov Models Oct 26th, 2023 Chengze Shen 1
Outline Multiple Sequence Alignment a. Background b. Profile Hidden Markov Models Getting An Alignment - Usage of Profile HMMs a. Single Profile HMM b. UPP c. WITCH Beyond profile HMMs - Usage of Progressive Alignment a. EMMA 2
Background - Multiple Sequence Alignment The input to Multiple Sequence Alignment (MSA) is a set of unaligned sequences (e.g., strings that consist of letters of nucleotides or amino acids). The output is an alignment of the sequences, adding gaps if necessary. 3
Background - Multiple Sequence Alignment To measure the accuracy of an MSA (compared to reference) Sum-of-pairs false negatives (SPFN) fraction of missed true homologies Sum-of-pairs false positives (SPFP) fraction of falsely recovered homologies 4
Background - Multiple Sequence Alignment MSA is a crucial step for many downstream biological analyses, such as a. Phylogeny estimation, detecting and quantifying selection, etc. b. Taxonomic identification/Abundance profiling Classic methods (e.g., MAFFT, MUSCLE, and Clustal-Omega). More recent methods (e.g., SATe, SATe-II, PASTA and MAGUS). a. Divide-and-conquer b. Align sub-problems with an accurate method (e.g., MAFFT-linsi) c. Merge sub-problems to form the final alignment d. Scalable to hundreds of thousands of sequences e. Accurate when rates of evolution are high 5
Background - Multiple Sequence Alignment However, only some methods try to deal with MSA with sequence length heterogeneity. Often appearing in biological datasets Also, if aligning reads/fragmentary sequences Additionally, many existing methods do not scale to aligning many sequences. E.g., aligning one million sequences General attempt to solve both limitations (two-stage approach) Align some sequences with an accurate method Add in the remaining sequences Figure 1 from the WITCH (Shen et al. 2023). Histograms of sequence lengths and x-axis refers to number of nucleotides (bp). 1000M1 is a simulated dataset; remaining datasets are biological and from Comparative Ribosome Website (CRW). 6
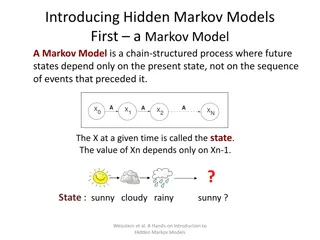
Background - Profile Hidden Markov Models Alignment as a profile HMM Given a profile HMM H and an unaligned sequence q, we can: Find the most probable path of q through H (Viterbi algorithm) Find the probability that H generates q (Forward algorithm) s t Figure credit: EMBL-EBI 7
Getting An Alignment - Use A Single Profile HMM (Recap) Multiple sequence alignment in two stages: a. Select a subset of sequences to be aligned (any method) b. Add in the remaining sequences Simplest idea: a. Use a single profile HMM to represent the subset alignment b. Add in the remaining sequences to the subset alignment by computing their most probable paths through the profile HMM AAAAATTTTTTAAAAA AAAAA----TTAAAAA AACAT----TTAAAA- Subset alignment -----AAAAATTTTTTAAAAA- -----AAAAA----TTAAAAA- -----AACAT----TTAAAA-- ccgga-AAAATTTT-------t New sequence CCGGAAAAATTTTT pHMM iiiiidmmmmmmmmdddddddi 8
Getting An Alignment - UPP A potential issue with using a single pHMM If an alignment has highly varied sequences (high rates of evolution), then the produced pHMM may poorly represent the alignment. Solution (UPP, Nguyen et al. 2015): Instead of creating a single pHMM, we break the alignment into smaller subsets Create a pHMM for each of the smaller subsets For each new sequence q: Find the most fitting pHMM that generates q and align it 9
Getting An Alignment - UPP 1. Separate unaligned sequences into two sets a. A subset of full-length sequences as backbone b. Remaining sequences as queries Estimate backbone alignment and tree Construct an ensemble of pHMMs (eHMM) to model the backbone alignment Assign each query sequence to a single best pHMM according to bit scores a. Then, align queries to their corresponding pHMMs Merge query alignments to backbone alignment with transitivity 2. 3. 4. Core idea: Representing the backbone with multiple pHMMs 5. Scoring metrics: HMMER (Finn et al. 2011) bit score (the fitness between a HMM H and a query sequence q). Figure 2 from UPP. 10
Getting An Alignment - UPP SPFN (Single pHMM) 11
Getting An Alignment - UPP Avg. over (1) 12.5%, (2) 25%, (3) 50% fragmentation 12
Getting An Alignment - WITCH WITCH (Shen et al. 2022b) improves upon UPP uses a different criterion to assign queries to HMMs (weighted bit scores) uses multiple HMMs instead of one HMM to align each query sequence WITCH New scoring metrics: weighted bit scores. 13
Improving the UPP Framework - WITCH 1. UPP selects the best scoring HMM model to align a sequence by bit scores. > Concern - Bit scores do not correctly reflect probabilities of query generation. > Improvement idea - replace bit scores with a more statistically rigorous calculation. 1. UPP utilizes one best HMM model to align a sequence. > Concern - the eHMM is underutilized, and there might be additional alignment information UPP ignores. > Improvement idea - align a sequence with multiple high-ranked HMMs and find a consensus alignment. q generated by Hx H1 Hx H2 Selected as the best HMM 14
pHMM Weighting Bit score Given Hi, how likely it generates q The definition of bit score is: Given a null/random model, how likely it generates q We define: siis the number of sequences in the alignment that generates Hi bit score between Hiand q 1. is the probability that HMM Higenerates sequence q. 1. Notice the impact of the number of sequences of the alignment that the HMM is built upon. 15
Finding A Consensus How to use the weighted pHMMs to align a sequence q? We can find a consensus alignment of q by combining alignments from multiple pHMMs: For a set of pHMMs {H1, H2, , Hn}: Alignment(q|H1)*w(H1,q) Alignment(q|H2)*w(H2,q) *In reality, only a few pHMMs may have impactful weights Alignment(q|H3)*w(H3,q) Alignment(q) = sum Alignment(q|Hn)*w(Hn,q ) 17
An Example of Consensus - Using 2 pHMMs Figure 3 from WITCH. An example of how WITCH combines multiple query-HMM alignments to form a consensus. The alignments can be merged by: Graph Clustering Merger (Smirnov and 1. Warnow 2021). 2. Maximum Weight Trace (Smirnov et al. 2022, Liu and Warnow 2023). 18
WITCH results - Datasets * p-distance is the normalized Hamming distance Testing datasets 19
WITCH results - High Fragmentary Datasets High fragmentation: 50% sequences are made to fragments of ~25% original lengths. Error: average of SPFN and SPFP MAGUS+UPP UPP when the backbone is aligned with MAGUS WITCH consistently improves on UPP on all datasets in terms of alignment error WITCH is the most accurate overall Partial Figure 5 from WITCH. Alignment error and Tree estimation false negative rates of methods. WITCH also leads to more accurate downstream tree estimations 20
WITCH results - High Fragmentary Datasets High fragmentation: 50% sequences are made to fragments of ~25% original lengths. Error: average of SPFN and SPFP Same story for the other model conditions a. But with lower rates of evolution (left to right), differences between methods are smaller Figure 5 from WITCH. Alignment error (top, average of SPFN and SPFP) and tree false negative rate (bottom, i.e., missing bipartitions) of WITCH and other methods on all sequences of simulated datasets with high fragmentation. 21
Conclusions - WITCH WITCH builds upon the UPP framework and introduces two algorithmic innovations: a. HMM weighting b. query consensus alignment with multiple weighted HMMs WITCH has improved alignment accuracy than UPP WITCH alignments yield more accurate maximum-likelihood tree estimation 24
Beyond profile HMM - Its Limitation pHMM-based method Advantage Disadvantage Fast and scales linearly to the number of queries Cannot find homologous pairs between queries that don t go through pHMM match states E.g., insertion states from two different aligned queries will never be aligned together 25
Beyond profile HMMs - Other Ways to Add Sequences Inputs: an existing alignment and a set of unaligned query sequences Output: an alignment on all sequences Method pHMM? Pros Cons MAFFT --add (progressive alignment) No Very fast, does not use HMMs Less accurate and memory hungry with a lot of sequences MAFFT-linsi --add (progressive alignment) No Accurate, does not use HMMs Very slow, memory hungry, and not scalable with more than 1000 sequences UPP Yes Accurate, fast and scalable to many sequences Query sequences are added independent to each other WITCH and WITCH-ng (same as WITCH but faster) Yes More accurate than UPP, also scalable to many sequences Same as UPP 26
Scaling Issue with MAFFT-linsi MAFFT-linsi --add more accurate than default MAFFT --add, but slower and less scalable. MAFFT-linsi failures due to OOM. Backbone: 1000-sequence alignment (dataset is INDELible 5000M2 averaged over 9 replicates). 28
EMMA pipeline 1. Build a tree on the existing alignment (subset alignment). Decompose the tree into subsets as in PASTA. subsets have induced sub-alignments Select subsets with L to U sequences. Construct HMMs on alignments of these selected subsets. 2. 3. 4. Modified Figure 1 from PASTA. 29
EMMA pipeline 1. Build a tree on the existing alignment (subset alignment). Decompose the tree into subsets as in PASTA. subsets have induced sub-alignments Select subsets with L to U sequences. Construct HMMs on alignments of these selected subsets. Assign query sequences to subsets based on adjusted HMM bit-scores. Align queries to alignments of the assigned subsets with MAFFT-linsi --add (progressive alignment instead of pHMMs). Merge all extended sub-alignments using transitivity. 2. 3. 4. 5. 6. Modified Figure 1 from PASTA. 7. Query sequences 30
EMMA - experiment results Backbone selection: 10% of all sequences, up to 1000 randomly selected sequences Remaining are queries Biological datasets X - failed to run # sequences 320 - 807 6,323 7,350 27,643 96,773 186,802 All methods generally have higher error on datasets with higher rates of evolution EMMA is the most accurate method 31
EMMA - experiment results Backbone selection: 10% of all sequences, up to 1000 randomly selected sequences Remaining are queries Simulated datasets X - failed to run # sequences 1000 1000 1000 1000 5000 5000 10,000 Similar story to biological data Bigger accuracy advantage for EMMA on datasets with high rates of evolution 32
Conclusions - EMMA EMMA successfully scales MAFFT-linsi --add to run on much larger datasets, including those up to ~186,000 sequences (Res) It usually has the best accuracy compared to WITCH-ng and even just MAFFT-linsi --add (when it can run) Comparing to HMM-based methods, EMMA can find homologies between added sequences 33