Understanding Chimeric Artifacts in Bacterial 16S rRNA Gene Sequencing
This content explores the formation of chimeric artifacts during PCR amplification of bacterial 16S rRNA gene segments, leading to challenges in biological sequence clustering and detection. It delves into the complexities of distinguishing between homologous and non-homologous crossover chimeras, providing insights on mitigating PCR-mediated recombination in molecular evolution studies using high-fidelity DNA polymerases.

Download Presentation

Please find below an Image/Link to download the presentation.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author. Download presentation by click this link. If you encounter any issues during the download, it is possible that the publisher has removed the file from their server.
E N D
Presentation Transcript
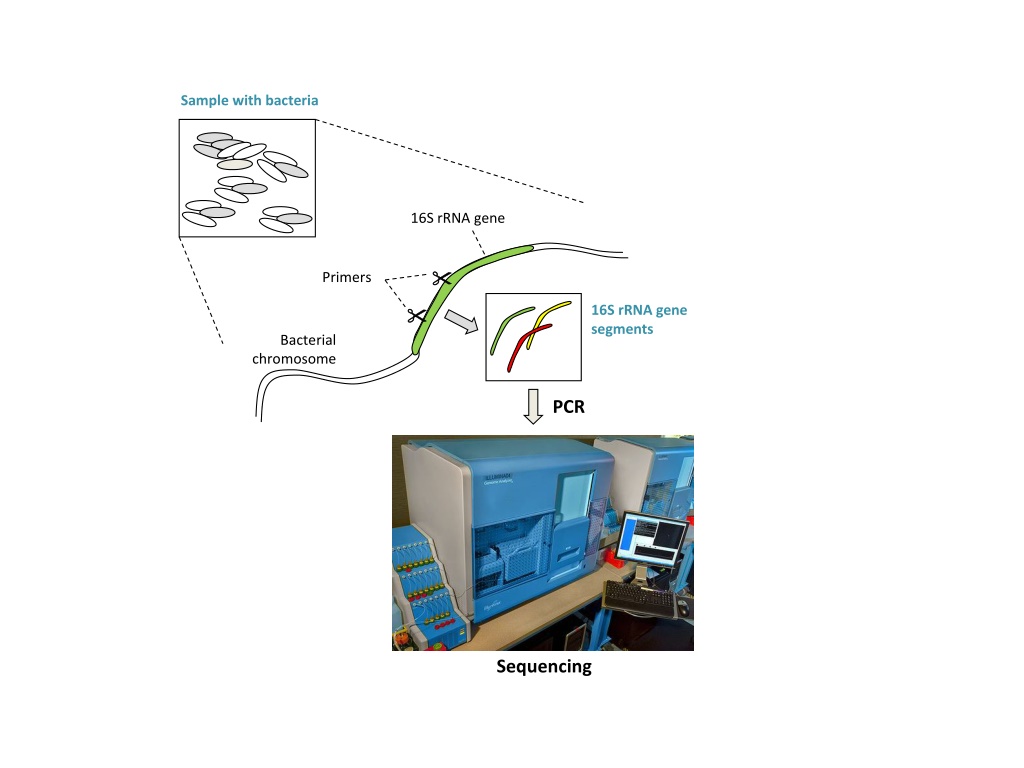
Sample with bacteria 16S rRNA gene Primers 16S rRNA gene segments Bacterial chromosome PCR Sequencing
Sample with bacteria 16S rRNA gene Primers 16S rRNA gene segments Bacterial chromosome PCR Amplified segments Biological sequences Sequencing Chimeric artifacts formed from 2 biological sequences during PCR
Biological OTUs Biological sequences Clustering Chimeric artifacts formed from 2 biological sequences during PCR Chimeric OTUs
From Haas et al. (2011) Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons, Genome Research.
Usually in region of high sequence similarity Similarity usually due to homology But not always! Non-homologous cross-over Chimera formed from single parent Looks like deletion or tandem duplication
Proportional to parent abundance Proportional to sequence similarity P(Parent1, Parent2) (proportional to) abundance(Parent1) abundance(Parent2) sequence_similarity(Parent1, Parent2) (cross-over at matching k-mer)
Bimera (two segments) Trimera (three segments) Trimera (three segs., two parents)
2-meras 3-meras 4-meras = (nr segments 1) From Lahr and Katz (2009) , Reducing the impact of PCR-mediated recombination in molecular evolution and environmental studies using a new-generation high-fidelity DNA polymerase, Biotechniques.
Homologous cross-over Chimeras look like biological sequences Often align well to reference sequences How to distinguish? Next-gen read 100 400nt 3% divergence = 3 12 diffs Small amount of evidence
Reference database Match segments to known parents De novo Find chimeric alignments (A-B-C) Chimera is least abundant UCHIME Edgar et al. (2001) UCHIME improves speed and sensitivity of chimera detection, Bioinformatics.
Haas et al. 2011. Find 50-mers unique to single genus Chimera if 50-mers indicate > 1 genus Low sensitivity, genus level only
Ashelford et al. 2005 At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl Environ Microbiol 71: 7724 7736 Find closest reference sequence(s) Measure divergence in sliding window (300nt) Compare with avg. variability in 16S gene Conserved vs. variable regions Anomaly (chimera) if variability far from avg. Newer algorithms work better
ChimeraSlayer Haas et al. 2011 Similar to UCHIME reference database mode Perseus Quince et al. 2011 Removing Noise From Pyrosequenced Amplicons ,BMC Bioinformatics. Similar to UCHIME de novo mode
Query Split into four chunks Chunk Chunk Chunk Chunk Search database Save top hits Hits Find & align closest pair (A, B) A Query B
User provides reference database Should be high-quality sequences Believed to be chimera-free Advantages: High confidence in predictions Disadvantages: Expect high false-negative rate Ref DB usually doesn't cover all possible parents
Parents amplified more than chimera At least one more round So parents at least 2x more abundant "Abundance skew" >= 2 (user-settable) Input is estimated amplicons + abundances NOT reads!
Sort amplicons by decreasing abundance Start with empty DB For each amplicon: Search DB for parents with >= 2x abundance If chimeric hit: Classify as chimera and discard query If not chimeric hit: Add to reference DB
Hits found by ref DB only (rare?) De novo Ref DB Hits found by de novo only (common? Hits found by both
Two modes check each other De novo should have better coverage All parents should be present Should examine hits found by ref DB but not by de novo See UCHIME manual for more discussion.
A 81 CCTTGGTAGGCCGtTGCCCTGCCAACTA GCTAATCAGACGC gggtCCATCtcaCACCaccggAgtTTTtcTCaCTgTacc 160 Q 81 CCTTGGTAGGCCGCTGCCCTGCCAACTA GCTAATCAGACGC ATCCCCATCCATCACCGATAAATCTTTAATCTCTTTCAG 160 B 81 TCTTGGTgGGCCGtTaCCCcGCCAACaA GCTAATCAGACGC ATCCCCATCCATCACCGATAAATCTTTAAaCTCTTTCAG 160 Diffs A A p A A A BBBB BBB BBBBB BB BBa B B BBB VotesY Y A Y Y Y YYYY YYY YYYYY YY YYN Y Y YYY Model AAAAAAAAAAAAAAAAAAAAAAAAAAAA xxxxxxxxxxxxx BBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBB Abstain vote No vote
A 81 CCTTGGTAGGCCGtTGCCCTGCCAACTA GCTAATCAGACGC gggtCCATCtcaCACCaccggAgtTTTtcTCaCTgTacc 160 Q 81 CCTTGGTAGGCCGCTGCCCTGCCAACTA GCTAATCAGACGC ATCCCCATCCATCACCGATAAATCTTTAATCTCTTTCAG 160 B 81 TCTTGGTgGGCCGtTaCCCcGCCAACaA GCTAATCAGACGC ATCCCCATCCATCACCGATAAATCTTTAAaCTCTTTCAG 160 Diffs A A p A A A BBBB BBB BBBBB BB BBa B B BBB VotesY Y A Y Y Y YYYY YYY YYYYY YY YYN Y Y YYY Model AAAAAAAAAAAAAAAAAAAAAAAAAAAA xxxxxxxxxxxxx BBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBB Model = segment of A + segment of B Chimeric if model closer to Q than A or B Left closer to A and right closer to B Closer if Y > N Ratio Y/N > 1
Observations: Y=Yes votes, N=No votes, A=abstain votes H= Y/ [ (N+n) +A] Parameters: =weight of No vote, n=prior number of No votes. Larger score = more likely to be chimera Default: chimera if HLeftx HRight 0.3 Threshold 0.3 adjusts sensitivity vs. false-positive rate.
Real communities Don't know all the biological sequences Can't distinguish chimeras from real (that's the problem!) Mock community Do you really know all the 16S sequences -- no! Communities too "easy" To few biological sequences, too well separated Simulation How realistic is it -- we don't know. No definitive validation
Noisy regions align well enough
Distribution of chimeric read fraction in 1247 samples (V3V5 and V6V4 ) Number of samples 0 5 10 15 20 25 30 Percent Chimeric
Chimera Detection Rate vs. GAST Distance 100% 90% 80% 70% Percent of Reads 60% 50% 40% 30% 20% 10% 0% 0 0.05 0.1 0.15 0.2 0.25 0.3 GAST Distance HMP V3-V5 HMP V6-V4 LSM V6-V4
Open-source version Source code donated to public domain USEARCH version Leverages proprietary algorithms 10x or more faster than open-source version
100x faster than Perseus 1,000x faster than ChimeraSlayer USEARCH version 10x or more faster again.
Many subtle issues Read manual & Supp. Mat. carefully!
Database incomplete Missing species Missing paralogs 16S duplications are common Probably high rates of false negatives
Parents should be present Probably low false negative rate vs. ref db. False positive rate not well known Mock community validation may be optimistic Error correction required Input MUST be amplicons & abundances Usually means starting from raw reads Cannot use on processed seqs. (e.g. RDP)
Convergent evolution in different clades Different rates in different regions Biological chimeras Bad sequences Bad alignments
Bad A Errors Good A Bad A Errors
Full-length gene Shotgun fragments Paired-end reads Reference database method De novo mode not possible with shotgun
Screen each end separately Using standard UCHIME in ref db. mode For each end E1,E2 Find closest parent P1,P2
P2 P2 Gap d1 using P1 P2 E2 E1 P1 Gap d2 using P2 NNNN... E1 E2 Pad gap with (d1 + d2)/2 Ns
UCHIME ref db. on padded pair. Ns don't count as diffs. NNNN... E1 E2
Ref db. mode can be run with any set of seqs. Later is more efficient (fewer seqs.) De novo mode no choice: Requires full set of amplicons & abundances Must follow denoise/error correction step De novo first Ref db. second Two modes can check each other
Based on USEARCH/UCLUST/UCHIME Described in afternoon talk on clustering.



 Chimeric 16S rRNA sequence formation")


")