Understanding Inborn Errors of Metabolism and Metabolic Disorders

Inborn Errors of Metabolism (IEM) are genetic disorders that disrupt metabolic pathways, leading to substrate accumulation or product deficiency. These disorders can be classified based on toxic accumulation, protein metabolism, carbohydrate intolerance, lysosomal storage issues, energy production defects, and more. Various examples of IEM include aminoacidopathies, urea cycle defects, disorders of carbohydrate metabolism, and lysosomal storage disorders. Identifying and understanding these conditions is crucial for early detection and effective management.

Download Presentation

Please find below an Image/Link to download the presentation.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author. Download presentation by click this link. If you encounter any issues during the download, it is possible that the publisher has removed the file from their server.
E N D
Presentation Transcript
Inborn Errors of Metabolism Dr Lucy Mungai University of Nairobi
Inborn Errors of Metabolism(IEM) Disorders in which a gene defect causes a clinically significant block in a metabolic pathway(enzyme deficiency or transport protein defect). Resulting either in accumulation of substrate before the block or deficiency of the product. Genetically transmitted
What is a metabolic disease? Garrod s hypothesis A C B product deficiency D Substrate excess toxic metabolite
Pathophysiologic classification Disorders that result in toxic accumulation Disorders of protein metabolism (eg, amino acidopathies, organic acidopathies, urea cycle defects) Disorders of carbohydrate intolerance Lysosomal storage disorders Disorders of energy production, utilization Fatty acid oxidation defects Disorders of carbohydrate utilization, production (ie, glycogen storage disorders, disorders of gluconeogenesis and glycogenolysis) Mitochondrial disorders Peroxisomal disorders
GLYCOGEN FAT PROTEIN FRUCTOSE GALACTOSE AMINO ACIDS GLUCOSE FREE FATTY ACIDS ORGANIC ACIDS AMMONIA PYRUVATE LACTATE ACETYL CoA UREA CYCLE KETONES UREA KREBS CYCLE NADH ATP An integrated view of the metabolic pathways
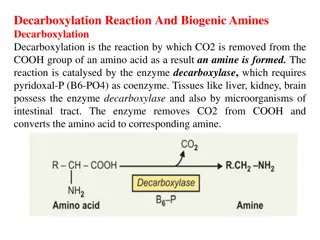
Classification of IEMs Aminoacidopathies e.g., phenylketonuria, hereditary tyrosinemia, nonketotic hyperglycinemia, maple syrup urine disease and homocystinuria. Hereditary tyrosinemia can present in the neonate with a bleeding diathesis due to liver disease, or later in infancy with a renal Fanconi syndrome. nonketotic hyperglycinemia presents as unremitting seizures with hypotonia and hiccoughs. MSUD classically presents at the end of the first week of life with feeding difficulties, lethargy, coma, seizures and the characteristic odor. Urea cycle defects (e.g., citrullinemia, ornithine transcarbamylase deficiency, and arginosuccinic aciduria) result from the inability to detoxify nitrogen and are characterized by severe hyperammonemia and respiratory alkalosis, with a typical onset after 24 hours of age
Classification of IEMs Disorders of carbohydrate metabolism (e.g., galactosemia, hereditary fructose intolerance, fructose 1,6-diphosphatase deficiency and the glycogen storage diseases) inability to metabolize specific sugars, aberrant glycogen synthesis, or disorders of gluconeogenesis. manifest with hypoglycemia, hepatosplenomegaly, lactic acidosis or ketosis. Lysosomal storage disorders (e.g., mucopolysaccharidosis, Tay-Sachs, Niemann-Pick disease, Gaucher s disease) caused by accumulation of glycoproteins, glycolipids, or glycosaminoglycans within lysosomes in various tissues. present later infancy, not with a specific laboratory abnormality, but with organomegaly, facial coarseness ,neurodegeneration and show a progressively degenerative course.
Classification of IEMs Organic acidemias (e.g. propionic acidemia, multiple carboxylase deficiency) abnormal metabolism of proteins, fats or carbohydrates with marked metabolic acidosis with ketosis, elevated lactate &hyperammonemia. Common signs include vomiting, signs of encephalopathy, neutropenia and thrombocytopenia. Fatty acid oxidation defects/Beta-oxidation defects e.g., short, medium, and long- chain acyl-CoA dehydrogenase deficiencies characterized by hypoketotic hypoglycemia, hyperammonemia & cardiomyopathy. (MCAD) is among the most common of all IEMs and may account for 5% of SIDS cases. Primary Lactic Acidoses present with severe lactic acidosis (e.g., pyruvate dehydrogenase, pyruvate carboxylase and cytochrome oxidase deficiencies).
Clinical Presentation Fetal development may have been normal, provided that the metabolites are able to cross the placenta and metabolized by mom. neonates may be asymptomatic, and may only become symptomatic after the initiation of feeds Newborn crash Multiple stillbirths or early childhood deaths suggestive, consanguinity unusual odor of urine or sweat
Clinical Presentation Toddlers and preschool-aged children present with stagnation or loss of cognitive milestones- language,attention developmental regression growth retardation seizures. History of growth disturbances, lethargy, recurrent emesis, poor feeding, rashes, seizures, hiccoughs, apnea, tachypnea.
History and Physical Subtle signs or symptoms Feeding difficulty, odd cry, vomiting, diarrhea, tachypnea, dyspnea, hypotonia/hypertonia, tachycardia, mental status changes Overt signs or symptoms Persistent hypoglycemia, acidosis, dehydration, shock, apnea, seizures, abnormal mental status, temperature instability, arrhythmia, cardiomyopathy, sudden death Dysmorphic features, strange odor, rashes, jaundice, organomegaly
Abnormal odors Burnt sugar,maple syrup MSUD Sweaty socks /cheese-like Isovaleric acidemia Fruity/ammoniacal Methylmalonic acidemia or propionic acidemia Mouse urine/musty Phenylketonuria Cabbage-like/rotten eggs Tyrosinemia Malt/hops Methionine malabsorption Cat urine 3-Methylcrotonic acidemia, 3-hydroxy-3- methylglutaric aciduria Fish-like Trimethylaminuria and carnitine excess
Physical Anomalies Associated With Acute-Onset Inborn Errors of Metabolism (IEM) Anomaly Ambiguous genitalia Hair and/or skin problems (alope- cia, dermatitis) Structural brain abnormalities (agenesis of corpus callosum, cortical cysts) Macrocephaly Renal cysts, facial dysmorphia Facial dysmorphia Cataract Retinopathy Lens dislocation, seizures Facial dysmorphia, congenital heart disease, vertebral anomalies Possible IEM Congentital adrenal hyperplasia Multiple carboxylase deficiency, biotinidase deficiency, argininosuccinic aciduria Pyruvate dehydrogenase deficiency Glutaric aciduria, type I Glutaric aciduria, type II; Zellweger syndrome Peroxisomal disorders, (Zellweger syndrome) Galactosemia, Lowe syndrome Peroxisomal disorders Sulfite oxidase deficiency Molybdenum cofactor deficiency 3-OH-isobutyric CoA deacylase deficiency
Inborn Errors of Metabolism of Acute Onset: Nonacidotic, Nonhyperammonemic Features Neurologic Features Predominant (Seizures, Hypotonia, Optic Abnormality) Glycine encephalopathy (nonketotic hyperglycinemia) Pyridoxine-responsive seizures Sulfite oxidase/santhine oxidase deficiency Peroxisomal disorders (Zellweger syndrome, neonatal adrenoleuko- dystrophy, infantile refsum disease) Jaundice Prominent Galactosemia Hereditary fructose intolerance Menkes kinky hair syndrome 1-antitrypsin deficiency Hypoglycemia (Nonketotic): Fatty acid oxidation defects (MCAD, LCAD, carnitine palmityl transferase, infantile form) Cardiomegaly Glycogen storage disease (type II phosphorylase kinase b deficiency18) Fatty acid oxidation defects (LCAD) Hepatomegaly (Fatty): Fatty acid oxidation defects (MCAD, LCAD) Skeletal Muscle Weakness: Fatty acid oxidation defects (LCAD, SCAD, multiple acyl-CoA dehydrogenase
Laboratory Assessment of Neonates Suspected of Having an Inborn Error of Metabolism Routine Studies Blood lactate and pyruvate Complete blood count and differential Plasma ammonia Plasma glucose Plasma electrolytes and blood pH Urine ketones Urine-reducing substances Special Studies Plasma amino acids Plasma carnitine Urine amino acids Urine organic acids
Laboratory findings Metabolic acidosis with increased anion gap, Primary respiratory alkalosis, hyperammonemia Hypoglycemia, ketosis or ketonuria, low BUN, Hyperbilirubinemia, elevated liver function tests including PT and PTT, lactic acidosis, high lactate/pyruvate ratio Non-glucose-reducing substances in urine, neutropenia and thrombocytopenia.
Approach to treatment Reduce precursor substrate load Removal of toxic compounds hemodialysis, chelators, trapping agents Enhancement of the activity of the deficient enzyme Decreasing the flux through the deficient pathway by restricting precursors in the diet. If an inborn error of metabolism is suspected in a neonate they should be kept NPO but given IV fluids containing dextrose so as to keep the infant anabolic. Provide caloric support Provide fluid support Divert metabolites Supplement with cofactor(s)
Hunter syndrome,mucopolysaccharidosis type 2 Mucopolysacchridoses disorder in breakdown of glycosaminoglycans(GAG) due to deficiency of lysosomal enzymes GAG- oligosaccharide components to protein skeleton in extracellular matrix Deficiency of the lysosomal enzyme iduronate-2- sulfatase (I2S) The lack of this enzyme causes heparan sulfate and dermatan sulfate to accumulate in all body tissues. Exhibit X-linked recessive inheritance
Epidemiology There are estimated to be approximately 2,000 people afflicted with Hunter syndrome worldwide, 500 of whom live in the United States. A study in the United Kingdom indicated an incidence among males of approximately 1 in 130,000 male live births. In severe cases, death usually occurs by age 15. In attenuated cases, patients may survive into their 50s
Clinical Presentation: Categorized as either "mild" or "severe" depending on the presence of central nervous system symptoms The symptoms not apparent at birth. As the buildup of GAGs continues throughout the cells of the body, signs of Hunter syndrome become more visible. distinctive coarseness in their facial features, prominent forehead, a nose with a flattened bridge, and an enlarged tongue. Communicating hydrocephalus can develop The gastrointestinal system is also affected via autonomic dysregulation , mucosal dysfunction.
Clinical Presentation: Valvular heart leaflets with GAG accumulation. A thickened myocardium eventually leads to coronary artery compromise, The walls of the airway may become thickened, as well, leading to obstructive airway disease. All major joints may be affected by Hunter syndrome, leading to joint stiffness and limited motion. carpal tunnel syndrome,short stature Pebbly, ivory-colored skin lesions may be found on the upper arms, legs, and upper back :pathognomic for the disease. After 18 months, children with severe MPS II may suffer from developmental decline
Investigations: Urine spot tests are readily available to screen for mucopolysaccharidoses (MPSs). ELISA technique has recently been shown to accurately quantify GAGs in urine (eg, dermatan and heparan sulfate excretion) and in blood. Lysosomal enzymes are present in all cells except mature erythrocytes. Iduronate sulfatase deficiency is determined by the direct enzymatic assay of leucocytes and fibroblasts as skin biopsy. Prenatal diagnosis is routinely available by measuring I2S enzymatic activity in amniotic fluid or in chorionic villus tissue A full skeletal survey Ophthalmologic examination with slit lamp: visual acuity and corneal and retinal . Cardiac echocardiography and ECG Airway evaluation: Assess for upper airway obstruction and sleep apnea and determine pulmonary function.[30
IEM- Index of Suspicion: Rapid deterioration in an otherwise well infant. Septic appearing infant or abnormal sepsis such as E.coli. Failure to thrive, Regression in milestones. Recurrent emesis or feeding difficulty, alterations in respirations, abnormal urine/body smell, changing MS/lethargy, jaundice, seizures, intractable hiccups. Can masquerade like pyloric stenosis. Dietary aversion- proteins, carbs.

")