NUTS AND BOLTS OF DFT CALCULATIONS


NUTS AND BOLTS OF DFT
NUTS AND BOLTS OF DFT
CALCULATIONS
CALCULATIONS

3.1 RECIPROCAL SPACE AND k POINTS
3.1 RECIPROCAL SPACE AND k POINTS
3.1.1 Plane Waves and the Brillouin Zone
3.1.1 Plane Waves and the Brillouin Zone
If we solve the Schrödinger equation for this periodic
If we solve the Schrödinger equation for this periodic
system, the solution must satisfy a fundamental property
system, the solution must satisfy a fundamental property
known as Bloch’s theorem, which states that the solution
known as Bloch’s theorem, which states that the solution
can be expressed as a sum of terms with the form:
can be expressed as a sum of terms with the form:
where u
where u
k
k
(r) is periodic in space with the same periodicity as
(r) is periodic in space with the same periodicity as
the supercell.
the supercell.
2
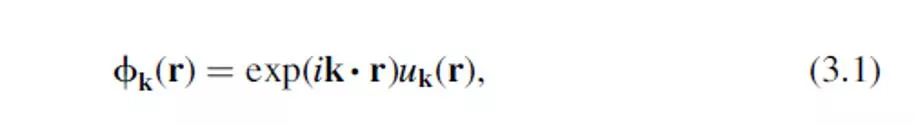
The space of vectors k is called reciprocal space (or simply k space).
The space of vectors k is called reciprocal space (or simply k space).
We define three vectors that define positions in reciprocal space.
We define three vectors that define positions in reciprocal space.
These vectors are called the reciprocal lattice vectors, b
These vectors are called the reciprocal lattice vectors, b
1
1
, b
, b
2
2
, and
, and
b
b
3
3
, and are defined so that a
, and are defined so that a
i
i
·b
·b
j
j
is 2
is 2
π
π
if i=j and 0 otherwise. This
if i=j and 0 otherwise. This
choice means that
choice means that
3
4
We can define a primitive cell in reciprocal space. Because
We can define a primitive cell in reciprocal space. Because
this cell has many special properties, it is given a name: it is
this cell has many special properties, it is given a name: it is
the Brillouin zone (often abbreviated to BZ).
the Brillouin zone (often abbreviated to BZ).
5
3.1.2 Choosing k Points in the Brillouin Zone
3.1.2 Choosing k Points in the Brillouin Zone
The solution that is used most widely was developed by
The solution that is used most widely was developed by
Monkhorst and Pack in 1976. Most DFT packages offer the
Monkhorst and Pack in 1976. Most DFT packages offer the
option of choosing k points based on this method.
option of choosing k points based on this method.
In practice, how should we choose how many k points to use?
In practice, how should we choose how many k points to use?
6
7
8
The last column in Table 3.2 lists the computational time
The last column in Table 3.2 lists the computational time
taken for the total energy calculations, normalized by the
taken for the total energy calculations, normalized by the
result for M=1.
result for M=1.
if M is an odd number, then the amount of time taken for the
if M is an odd number, then the amount of time taken for the
calculations with either M or (M+1) was close to the same.
calculations with either M or (M+1) was close to the same.
This occurs because the calculations take full advantage of the
This occurs because the calculations take full advantage of the
many
many
symmetries
symmetries
that exist in a perfect fcc solid.
that exist in a perfect fcc solid.
9
This reduced region in k space is called the
This reduced region in k space is called the
irreducible
irreducible
Brillouin zone (IBZ)
Brillouin zone (IBZ)
.
.
To show how helpful symmetry is in reducing the work
To show how helpful symmetry is in reducing the work
required for a DFT calculation, we have repeated some of
required for a DFT calculation, we have repeated some of
the calculations from Table 3.2 for a four-atom supercell in
the calculations from Table 3.2 for a four-atom supercell in
which each atom was given a slight displacement away
which each atom was given a slight displacement away
from its fcc lattice position.
from its fcc lattice position.
10
11
3.1.3 Metals—Special Cases in k Space
3.1.3 Metals—Special Cases in k Space
One useful definition of a metal is that in a metal the Brillouin
One useful definition of a metal is that in a metal the Brillouin
zone can be divided into regions that are occupied and
zone can be divided into regions that are occupied and
unoccupied by electrons. The surface in k space that separates
unoccupied by electrons. The surface in k space that separates
these two regions is called the
these two regions is called the
Fermi surface
Fermi surface
.
.
12
We will describe the two best-known methods to solve the
We will describe the two best-known methods to solve the
problem of slow convergence of metal calculations.
problem of slow convergence of metal calculations.
The first is called
The first is called
the tetrahedron method
the tetrahedron method
.
.
The idea behind this method is
The idea behind this method is
to use the discrete set of k
to use the discrete set of k
points to define a set of tetrahedra that fill reciprocal space
points to define a set of tetrahedra that fill reciprocal space
and to define the function being integrated at every point in a
and to define the function being integrated at every point in a
tetrahedron using interpolation.
tetrahedron using interpolation.
13
A different approach to the discontinuous integrals that
A different approach to the discontinuous integrals that
appear for metals are
appear for metals are
the smearing methods.
the smearing methods.
The idea of these methods is
The idea of these methods is
to force the function being
to force the function being
integrated to be continuous by “smearing” out the
integrated to be continuous by “smearing” out the
discontinuity.
discontinuity.
14
An example of a smearing function is the Fermi–Dirac
An example of a smearing function is the Fermi–Dirac
function:
function:
Figure 3.3 shows the shape of this function for several
Figure 3.3 shows the shape of this function for several
values of
values of
σ
σ
.
.
15
16
3.1.4 Summary of k Space
3.1.4 Summary of k Space
The key ideas related to getting well-converged results in k
The key ideas related to getting well-converged results in k
space include:
space include:
1. Before pursuing a large series of DFT calculations for a
1. Before pursuing a large series of DFT calculations for a
system of interest, numerical data exploring the
system of interest, numerical data exploring the
convergence of the calculations with respect to the number
convergence of the calculations with respect to the number
of k points should be obtained.
of k points should be obtained.
2. The number of k points used in any calculation should be
2. The number of k points used in any calculation should be
reported since not doing so makes reproduction of the
reported since not doing so makes reproduction of the
result difficult.
result difficult.
17
3. Increasing the volume of a supercell reduces the number
3. Increasing the volume of a supercell reduces the number
of k points needed to achieve convergence because volume
of k points needed to achieve convergence because volume
increases in real space correspond to volume decreases in
increases in real space correspond to volume decreases in
reciprocal space.
reciprocal space.
4. If calculations involving supercells with different volumes
4. If calculations involving supercells with different volumes
are to be compared, choosing k points so that the density
are to be compared, choosing k points so that the density
of k points in reciprocal space is comparable for the
of k points in reciprocal space is comparable for the
different supercells is a useful way to have comparable
different supercells is a useful way to have comparable
levels of convergence in k space.
levels of convergence in k space.
18
5. Understanding how symmetry is used to reduce the
5. Understanding how symmetry is used to reduce the
number of k points for which calculations are actually
number of k points for which calculations are actually
performed can help in understanding how long individual
performed can help in understanding how long individual
calculations will take. But overall convergence is
calculations will take. But overall convergence is
determined by the density of k points in the full Brillouin
determined by the density of k points in the full Brillouin
zone, not just the number of k points in the irreducible
zone, not just the number of k points in the irreducible
Brillouin zone.
Brillouin zone.
6. Appropriate methods must be used to accurately treat k
6. Appropriate methods must be used to accurately treat k
space for metals.
space for metals.
19
3.2 ENERGY CUTOFFS
3.2 ENERGY CUTOFFS
Our lengthy discussion of k space began with Bloch’s
Our lengthy discussion of k space began with Bloch’s
theorem, which tells us that solutions of the Schrödinger
theorem, which tells us that solutions of the Schrödinger
equation for a supercell have the form
equation for a supercell have the form
where u
where u
k
k
(r) is periodic in space with the same periodicity as
(r) is periodic in space with the same periodicity as
the supercell. It is now time to look at this part of the
the supercell. It is now time to look at this part of the
problem more carefully. The periodicity of u
problem more carefully. The periodicity of u
k
k
(r) means that
(r) means that
it can be expanded in terms of a special set of plane waves:
it can be expanded in terms of a special set of plane waves:
20
Combining the two equations above gives:
Combining the two equations above gives:
The functions appearing in Eq.(3.13) have a simple
The functions appearing in Eq.(3.13) have a simple
interpretation as solutions of the Schrödinger equation:
interpretation as solutions of the Schrödinger equation:
they are solutions with kinetic energy:
they are solutions with kinetic energy:
21
As a result, it is usual to truncate the infinite sum above to
As a result, it is usual to truncate the infinite sum above to
include only solutions with kinetic energies less than some
include only solutions with kinetic energies less than some
value:
value:
The infinite sum then reduces to :
The infinite sum then reduces to :
22
23
3.2.1 Pseudopotentials
3.2.1 Pseudopotentials
A pseudopotential replaces the electron density
A pseudopotential replaces the electron density
from a chosen set of core electrons with a smoothed
from a chosen set of core electrons with a smoothed
density chosen to match various important physical
density chosen to match various important physical
and mathematical properties of the true ion core.
and mathematical properties of the true ion core.
24
A pseudopotential is developed by considering an
A pseudopotential is developed by considering an
isolated atom of one element, but the resulting
isolated atom of one element, but the resulting
pseudopotential can then
pseudopotential can then
be used reliably for
be used reliably for
calculations that place this atom in any chemical
calculations that place this atom in any chemical
environment
environment
without further adjustment
without further adjustment
of the
of the
pseudopotential.
pseudopotential.
25
3.3 NUMERICAL OPTIMIZATION
3.3 NUMERICAL OPTIMIZATION
To make practical use of our ability to perform numerically
To make practical use of our ability to perform numerically
converged DFT calculations, we also need methods that can
converged DFT calculations, we also need methods that can
help us effectively cope with situations where we want to
help us effectively cope with situations where we want to
search through a problem with many degrees of freedom.
search through a problem with many degrees of freedom.
26
3.3.1 Optimization in One Dimension
3.3.1 Optimization in One Dimension
How to find a local minimum of f (x)?
How to find a local minimum of f (x)?
The first approach is the
The first approach is the
bisection method
bisection method
.
.
The second approach is
The second approach is
Newton’s method
Newton’s method
.
.
27
28
A striking difference between the bisection method and
A striking difference between the bisection method and
Newton’s method is how rapidly the solutions converge.
Newton’s method is how rapidly the solutions converge.
29
Newton’s method is better than the bisection method
Newton’s method is better than the bisection method
How do we know when to stop?
How do we know when to stop?
A typical choice is to continue iterating until the
A typical choice is to continue iterating until the
difference between successive iterates is smaller
difference between successive iterates is smaller
than some tolerance:
than some tolerance:
30
there are several general properties of numerical
there are several general properties of numerical
optimization that are extremely important to appreciate.
optimization that are extremely important to appreciate.
They include :
They include :
1. The algorithms are iterative, so they do not provide an
1. The algorithms are iterative, so they do not provide an
exact solution; instead, they provide a series of
exact solution; instead, they provide a series of
approximations to the exact solution.
approximations to the exact solution.
2. An initial estimate for the solution must be provided to
2. An initial estimate for the solution must be provided to
use the algorithms.The algorithms provide no guidance on
use the algorithms.The algorithms provide no guidance on
how to choose this initial estimate.
how to choose this initial estimate.
31
3. The number of iterations performed is controlled by a
3. The number of iterations performed is controlled by a
tolerance parameter that estimates how close the current
tolerance parameter that estimates how close the current
solution is to the exact solution.
solution is to the exact solution.
4. Repeating any algorithm with a different tolerance
4. Repeating any algorithm with a different tolerance
parameter or a different initial estimate for the solution will
parameter or a different initial estimate for the solution will
generate multiple final approximate solutions that are
generate multiple final approximate solutions that are
(typically) similar but are not identical.
(typically) similar but are not identical.
5. The rate at which different algorithms converge to a
5. The rate at which different algorithms converge to a
solution can vary greatly, so choosing an appropriate
solution can vary greatly, so choosing an appropriate
algorithm can greatly reduce the number of iterations
algorithm can greatly reduce the number of iterations
needed.
needed.
32
6. These methods cannot tell us if there are multiple minima
6. These methods cannot tell us if there are multiple minima
of the function we are considering if we just apply the
of the function we are considering if we just apply the
method once. Applying a method multiple times with
method once. Applying a method multiple times with
different initial estimates can yield multiple minima, but
different initial estimates can yield multiple minima, but
even in this case the methods do not give enough
even in this case the methods do not give enough
information to prove that all possible minima have been
information to prove that all possible minima have been
found.
found.
7. For most methods, no guarantees can be given that the
7. For most methods, no guarantees can be given that the
method will converge to a solution at all for an arbitrary
method will converge to a solution at all for an arbitrary
initial estimate.
initial estimate.
33
3.3.2 Optimization in More than One Dimension
3.3.2 Optimization in More than One Dimension
The one-dimensional Newton method was derived using a
The one-dimensional Newton method was derived using a
Taylor expansion, and the multidimensional problem can be
Taylor expansion, and the multidimensional problem can be
approached in the same way.
approached in the same way.
Newton’s method defines a series of iterates by:
Newton’s method defines a series of iterates by:
34
Unfortunately, Newton’s method simply cannot be
Unfortunately, Newton’s method simply cannot be
applied to the DFT problem we set ourselves at the
applied to the DFT problem we set ourselves at the
beginning of this section!
beginning of this section!
35
It is very difficult to directly evaluate second
It is very difficult to directly evaluate second
derivatives of energy within plane-wave DFT, and
derivatives of energy within plane-wave DFT, and
most codes do not attempt to perform these
most codes do not attempt to perform these
calculations.
calculations.
36
As a result, we have to look for other approaches to
As a result, we have to look for other approaches to
minimize E(x). We will briefly discuss the two
minimize E(x). We will briefly discuss the two
numerical methods that are most commonly used
numerical methods that are most commonly used
for this problem:
for this problem:
quasi-Newton
quasi-Newton
and
and
conjugate-
conjugate-
gradient methods
gradient methods
.
.
37
The essence of
The essence of
quasi-Newton methods
quasi-Newton methods
is to replace Eq.
is to replace Eq.
(3.15) by :
(3.15) by :
where A
where A
i
i
is a matrix that is defined to approximate the
is a matrix that is defined to approximate the
Jacobian matrix. This matrix is also updated iteratively
Jacobian matrix. This matrix is also updated iteratively
during the calculation and has the form:
during the calculation and has the form:
38
Conjugate gradient method:
Conjugate gradient method:
We begin with an initial estimate, x
We begin with an initial estimate, x
0
0
. Our first iterate is
. Our first iterate is
chosen to lie along the direction defined by
chosen to lie along the direction defined by
the problem of choosing the best value of
the problem of choosing the best value of
α
α
0
0
cannot be
cannot be
solved exactly. Choose the step size by some approximate
solved exactly. Choose the step size by some approximate
method that may be as simple as evaluating E
method that may be as simple as evaluating E
(x1)
(x1)
for several
for several
possible step lengths and selecting the best result.
possible step lengths and selecting the best result.
39
3
3
.
.
4
4
G
G
E
E
O
O
M
M
E
E
T
T
R
R
Y
Y
O
O
P
P
T
T
I
I
M
M
I
I
Z
Z
A
A
T
T
I
I
O
O
N
N
3.4.1 Internal Degrees of Freedom
3.4.1 Internal Degrees of Freedom
Example 1 : Obtain the energy of the gas phase N
Example 1 : Obtain the energy of the gas phase N
2
2
molecule
molecule
Step 1 :Create a cubic super-cell with a side length of L
Step 1 :Create a cubic super-cell with a side length of L
Step 2 :Place two N atoms at the fractional coordinates (0,
Step 2 :Place two N atoms at the fractional coordinates (0,
0, 0) and ( + d/L , O , 0) of this supercell.
0, 0) and ( + d/L , O , 0) of this supercell.
Step 3:find the DFT-optimized bond length for N
Step 3:find the DFT-optimized bond length for N
2
2
by using
by using
either the quasi-Newton or conjugate-gradient methods
either the quasi-Newton or conjugate-gradient methods
defined above to minimize the total energy of our supercell.
defined above to minimize the total energy of our supercell.
40
Example 2 : Optimize the geometry of a molecule of CO
Example 2 : Optimize the geometry of a molecule of CO
2
2
Step 1 :Create a cubic super-cell with a side length of L
Step 1 :Create a cubic super-cell with a side length of L
Step 2 :Create a CO2 molecule by placing a C atom at
Step 2 :Create a CO2 molecule by placing a C atom at
fractional coordinates (0,0,0) and O atoms at (+d/L,0,0) and
fractional coordinates (0,0,0) and O atoms at (+d/L,0,0) and
(-d/L,0,0).
(-d/L,0,0).
Step 3:find the DFT-optimized bond length for CO
Step 3:find the DFT-optimized bond length for CO
2
2
by using
by using
either the quasi-Newton or conjugate-gradient methods
either the quasi-Newton or conjugate-gradient methods
defined above to minimize the total energy of our supercell.
defined above to minimize the total energy of our supercell.
41
3.4.2 Geometry Optimization with Constrained
3.4.2 Geometry Optimization with Constrained
Atoms
Atoms
There are many types of calculations where it is useful to
There are many types of calculations where it is useful to
minimize the energy of a supercell by optimizing the
minimize the energy of a supercell by optimizing the
position of some atoms while holding other atoms at fixed
position of some atoms while holding other atoms at fixed
positions.
positions.
42
Exploring the fundamental concepts of DFT calculations including reciprocal space, Brillouin zone, k points selection, and computational efficiency. Learn about Bloch's theorem, Monkhorst-Pack method, and the importance of symmetry in reducing computational workload. Dive into the world of DFT with this comprehensive guide.

Download Presentation

Please find below an Image/Link to download the presentation.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author.If you encounter any issues during the download, it is possible that the publisher has removed the file from their server.
You are allowed to download the files provided on this website for personal or commercial use, subject to the condition that they are used lawfully. All files are the property of their respective owners.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author.
E N D
Presentation Transcript
NUTS AND BOLTS OF DFT CALCULATIONS
3.1 RECIPROCAL SPACE AND k POINTS 3.1.1 Plane Waves and the Brillouin Zone If we solve the Schr dinger equation for this periodic system, the solution must satisfy a fundamental property known as Bloch s theorem, which states that the solution can be expressed as a sum of terms with the form: where uk(r) is periodic in space with the same periodicity as the supercell. 2
The space of vectors k is called reciprocal space (or simply k space). We define three vectors that define positions in reciprocal space. These vectors are called the reciprocal lattice vectors, b1, b2, and b3, and are defined so that ai bjis 2 if i=j and 0 otherwise. This choice means that 3
We can define a primitive cell in reciprocal space. Because this cell has many special properties, it is given a name: it is the Brillouin zone (often abbreviated to BZ). 5
3.1.2 Choosing k Points in the Brillouin Zone The solution that is used most widely was developed by Monkhorst and Pack in 1976. Most DFT packages offer the option of choosing k points based on this method. In practice, how should we choose how many k points to use? 6
The last column in Table 3.2 lists the computational time taken for the total energy calculations, normalized by the result for M=1. if M is an odd number, then the amount of time taken for the calculations with either M or (M+1) was close to the same. This occurs because the calculations take full advantage of the many symmetries that exist in a perfect fcc solid. 9
This reduced region in k space is called the irreducible Brillouin zone (IBZ). To show how helpful symmetry is in reducing the work required for a DFT calculation, we have repeated some of the calculations from Table 3.2 for a four-atom supercell in which each atom was given a slight displacement away from its fcc lattice position. 10
3.1.3 MetalsSpecial Cases in k Space One useful definition of a metal is that in a metal the Brillouin zone can be divided into regions that are occupied and unoccupied by electrons. The surface in k space that separates these two regions is called the Fermi surface. 12
We will describe the two best-known methods to solve the problem of slow convergence of metal calculations. The first is called the tetrahedron method. The idea behind this method is to use the discrete set of k points to define a set of tetrahedra that fill reciprocal space and to define the function being integrated at every point in a tetrahedron using interpolation. 13
A different approach to the discontinuous integrals that appear for metals are the smearing methods. The idea of these methods is to force the function being integrated to be continuous discontinuity. by smearing out the 14
An example of a smearing function is the FermiDirac function: Figure 3.3 shows the shape of this function for several values of . 15
3.1.4 Summary of k Space The key ideas related to getting well-converged results in k space include: 1. Before pursuing a large series of DFT calculations for a system of interest, numerical convergence of the calculations with respect to the number of k points should be obtained. 2. The number of k points used in any calculation should be reported since not doing so makes reproduction of the result difficult. data exploring the 17
3. Increasing the volume of a supercell reduces the number of k points needed to achieve convergence because volume increases in real space correspond to volume decreases in reciprocal space. 4. If calculations involving supercells with different volumes are to be compared, choosing k points so that the density of k points in reciprocal space is comparable for the different supercells is a useful way to have comparable levels of convergence in k space. 18
5. Understanding how symmetry is used to reduce the number of k points for which calculations are actually performed can help in understanding how long individual calculations will take. But determined by the density of k points in the full Brillouin zone, not just the number of k points in the irreducible Brillouin zone. 6. Appropriate methods must be used to accurately treat k space for metals. overall convergence is 19
3.2 ENERGY CUTOFFS Our lengthy discussion of k space began with Bloch s theorem, which tells us that solutions of the Schr dinger equation for a supercell have the form where uk(r) is periodic in space with the same periodicity as the supercell. It is now time to look at this part of the problem more carefully. The periodicity of uk(r) means that it can be expanded in terms of a special set of plane waves: 20
Combining the two equations above gives: The interpretation as solutions of the Schr dinger equation: they are solutions with kinetic energy: functions appearing in Eq.(3.13) have a simple 21
As a result, it is usual to truncate the infinite sum above to include only solutions with kinetic energies less than some value: The infinite sum then reduces to : 22
3.2.1 Pseudopotentials A pseudopotential replaces the electron density from a chosen set of core electrons with a smoothed density chosen to match various important physical and mathematical properties of the true ion core. 24
A pseudopotential is developed by considering an isolated atom of one element, but the resulting pseudopotential can then be calculations that place this atom in any chemical environment without further adjustment of the pseudopotential. used reliably for 25
3.3 NUMERICAL OPTIMIZATION To make practical use of our ability to perform numerically converged DFT calculations, we also need methods that can help us effectively cope with situations where we want to search through a problem with many degrees of freedom. 26
3.3.1 Optimization in One Dimension How to find a local minimum of f (x)? The first approach is the bisection method. The second approach is Newton s method. 27
A striking difference between the bisection method and Newton s method is how rapidly the solutions converge. Newton s method is better than the bisection method 29
How do we know when to stop? A typical choice is to continue iterating until the difference between successive iterates is smaller than some tolerance: 30
there optimization that are extremely important to appreciate. They include : 1. The algorithms are iterative, so they do not provide an exact solution; instead, they approximations to the exact solution. 2. An initial estimate for the solution must be provided to use the algorithms.The algorithms provide no guidance on how to choose this initial estimate. are several general properties of numerical provide a series of 31
3. The number of iterations performed is controlled by a tolerance parameter that estimates how close the current solution is to the exact solution. 4. Repeating any algorithm with a different tolerance parameter or a different initial estimate for the solution will generate multiple final approximate solutions that are (typically) similar but are not identical. 5. The rate at which different algorithms converge to a solution can vary greatly, so choosing an appropriate algorithm can greatly reduce the number of iterations needed. 32
6. These methods cannot tell us if there are multiple minima of the function we are considering if we just apply the method once. Applying a method multiple times with different initial estimates can yield multiple minima, but even in this case the methods do not give enough information to prove that all possible minima have been found. 7. For most methods, no guarantees can be given that the method will converge to a solution at all for an arbitrary initial estimate. 33
3.3.2 Optimization in More than One Dimension The one-dimensional Newton method was derived using a Taylor expansion, and the multidimensional problem can be approached in the same way. Newton s method defines a series of iterates by: 34
Unfortunately, Newtons method simply cannot be applied to the DFT problem we set ourselves at the beginning of this section! 35
It is very difficult to directly evaluate second derivatives of energy within plane-wave DFT, and most codes do not attempt to perform these calculations. 36
As a result, we have to look for other approaches to minimize E(x). We will briefly discuss the two numerical methods that are most commonly used for this problem: quasi-Newton and conjugate- gradient methods. 37
The essence of quasi-Newton methods is to replace Eq. (3.15) by : where Aiis a matrix that is defined to approximate the Jacobian matrix. This matrix is also updated iteratively during the calculation and has the form: 38
Conjugate gradient method: We begin with an initial estimate, x0. Our first iterate is chosen to lie along the direction defined by the problem of choosing the best value of 0cannot be solved exactly. Choose the step size by some approximate method that may be as simple as evaluating E(x1)for several possible step lengths and selecting the best result. 39
3.4 GEOMETRY OPTIMIZATION 3.4.1 Internal Degrees of Freedom Example 1 : Obtain the energy of the gas phase N2 molecule Step 1 :Create a cubic super-cell with a side length of L Step 2 :Place two N atoms at the fractional coordinates (0, 0, 0) and ( + d/L , O , 0) of this supercell. Step 3:find the DFT-optimized bond length for N2by using either the quasi-Newton or conjugate-gradient methods defined above to minimize the total energy of our supercell. 40
Example 2 : Optimize the geometry of a molecule of CO2 Step 1 :Create a cubic super-cell with a side length of L Step 2 :Create a CO2 molecule by placing a C atom at fractional coordinates (0,0,0) and O atoms at (+d/L,0,0) and (-d/L,0,0). Step 3:find the DFT-optimized bond length for CO2by using either the quasi-Newton or conjugate-gradient methods defined above to minimize the total energy of our supercell. 41
3.4.2 Atoms Geometry Optimization with Constrained There are many types of calculations where it is useful to minimize the energy of a supercell by optimizing the position of some atoms while holding other atoms at fixed positions. 42