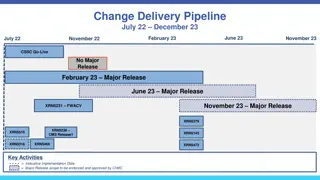
Genomic Imputation Pipeline Overview


WHI Imputation

Target GWAS data
•
WHIMS +, ~5,000-6,000 samples, Illumina Omni
express
•
GRANET, ~5,000 samples, Illumina Omni
•
Hipfx, ~4,000-5,000 samples, Illumina 550k-610k
•
SHARE, ~12,000 samples, Affy 6 (already imputed)
•
GECCO, ~4,000 samples,
Illumina 550K, 550K duo,
610K, 300K (already imputed)
•
WHI NCI GWAS, ~1000 samples, not cleaned and will
not be included in the current batch of imputations
Reference Panel
•
Reference Panel: 1000G Phase I Integrated
Release Version 2 Haplotypes; downloaded
from MACH website
•
1092 samples (
AFR 246;AMR 181;ASN 286;
EUR 379
). We used all population because it
has been shown to increase the imputation
quality of the rare variants.
•
36,648,992 SNPs; 3,660,720 indels
Imputation steps
•
Convert the GWAS data from HG18 to 19
•
Matched the strand in GWAS data with 1000
genomes reference panel (forward strand)
•
Phased each study separately using Beagle
•
Impute each study using minimac. Will break
chromosomes small chunks to facilitate the
imputation process; there will be overlapping
between neighbouring chunks, which
improves accuracy near the edges
Expected output
•
The imputation program will output the imputed dosage and the
imputation quality measure R2 for each SNP.
•
Previously in imputation of GECCO data, we determined the R2
cutoff by MAF categories to filter SNPs
1.
For MAF>0.01: R2>0.3
2.
For 0.005<MAF<0.01: R2>0.5
3.
For MAF<0.005: R2>0.99
•
Around 9M SNPs and 1.2M indels left after the filtering
•
Comparison of SNPs that pass the filtering for Hapmap and 1000
genomes imputation
Timeline
•
It will take approximately one half month to
finish the imputation for each study
•
So the imputation should be done by the end
of May 2013 assuming we get access to GWAS
data in a timely manner.
This document outlines a genomic imputation pipeline for multiple GWAS studies using reference panels such as 1000 Genomes Phase I data. It covers steps like data matching, phasing, and imputation using tools like Beagle and Minimac. The expected output includes imputed dosages and quality measures. Filtering criteria based on minor allele frequency are defined, and a timeline is provided for completing the imputation by the end of May 2013.

Download Presentation

Please find below an Image/Link to download the presentation.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author.If you encounter any issues during the download, it is possible that the publisher has removed the file from their server.
You are allowed to download the files provided on this website for personal or commercial use, subject to the condition that they are used lawfully. All files are the property of their respective owners.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author.
E N D
Presentation Transcript
Target GWAS data WHIMS +, ~5,000-6,000 samples, Illumina Omni express GRANET, ~5,000 samples, Illumina Omni Hipfx, ~4,000-5,000 samples, Illumina 550k-610k SHARE, ~12,000 samples, Affy 6 (already imputed) GECCO, ~4,000 samples, Illumina 550K, 550K duo, 610K, 300K (already imputed) WHI NCI GWAS, ~1000 samples, not cleaned and will not be included in the current batch of imputations
Reference Panel Reference Panel: 1000G Phase I Integrated Release Version 2 Haplotypes; downloaded from MACH website 1092 samples (AFR 246;AMR 181;ASN 286; EUR 379). We used all population because it has been shown to increase the imputation quality of the rare variants. 36,648,992 SNPs; 3,660,720 indels
Imputation steps Convert the GWAS data from HG18 to 19 Matched the strand in GWAS data with 1000 genomes reference panel (forward strand) Phased each study separately using Beagle Impute each study using minimac. Will break chromosomes small chunks to facilitate the imputation process; there will be overlapping between neighbouring chunks, which improves accuracy near the edges
Expected output The imputation program will output the imputed dosage and the imputation quality measure R2 for each SNP. Previously in imputation of GECCO data, we determined the R2 cutoff by MAF categories to filter SNPs 1. For MAF>0.01: R2>0.3 2. For 0.005<MAF<0.01: R2>0.5 3. For MAF<0.005: R2>0.99 Around 9M SNPs and 1.2M indels left after the filtering Comparison of SNPs that pass the filtering for Hapmap and 1000 genomes imputation MAF<0.5% [0.5%, 1%) >1% Hapmap 287 70,695 720,567 1000 genomes 4,532 875,658 9,309,439
Timeline It will take approximately one half month to finish the imputation for each study So the imputation should be done by the end of May 2013 assuming we get access to GWAS data in a timely manner.